Discusión del caso clínico. | Presentación |
Se nos presenta el caso de un paciente varón de 47 años, sin antecedentes de jerarquía, que inicia con un cuadro progresivo de lesiones cutáneas hipocrómicas asociadas a cefalea y trastornos visuales, evidenciándose en TAC de cráneo la presencia de un tumor supraselar.
En primer lugar planteo cuáles son los tumores de localización supraselar más frecuentes y si su presencia puede justificar los síntomas de nuestro paciente, incluídas las lesiones cutáneas, o si estas pueden obedecer a otras causas como ser fenómenos autoinmunes, neurocitotóxicos o medioambientales como se plantea en los casos de vitíligo. Por otro lado me pregunto qué jerarquía adquiriría el vitíligo como dermatosis paraneoplásica en nuestro paciente y finalmente cuál sería el abordaje terapéutico a seguir en este caso.
Los tumores de localización supraselar son aquellos que se encuentran por encima de la silla turca, entre el área de la hipófisis, el infundíbulo y el ventrículo medio. Los más frecuentes son: los gliomas del quiasma óptico, los meningiomas y el craneofaringioma.
• Glioma del quiasma óptico : es el más frecuente en niños y se maniefiesta por disminución de la agudeza visual, disfunción endócrina y síndrome diencefálico. Se presenta en un 20 a 50% de pacientes con neurifibromatosis tipo I. En la resonancia magnética (RMN) de cráneo puede visualizarse como una lesión isointensa en T1 con refuerzo heterogéneo tras la administración de contraste.
• Meningioma : predomina en el adulto. Son tumores benignos, bien delimitados y de crecimiento lento. En el 8% de los casos se localizan en la región supraselar y la sintomatología depende de la localización y la extensión de los mismos pudiendo presentarse con síntomas neurológicos inespecíficos o focales. En los estudios por imágenes, principalmente la RMN presentan un importante realce tras la administración del contraste
• Craneofaringioma : puede presentarse tanto en la niñez como en la edad adulta. Si bien es un tumor benigno, puede tener un gran crecimiento y adoptar un comportamiento agresivo local. En la RMN suele ser característico la presencia de un componente quístico o quístico/sólido con calcificaciones presentando alto realce con el contraste. Por este motivo considero que, al menos por las características a nivel de imágenes, podríamos estar ante un craneofaringioma.
Craneofaringioma.
Es un tumor de aspecto quístico o quístico – sólido que deriva de remanentes de células embrionarias de la bolsa de Rathke. Son de carácter benigno y de lento crecimiento pero con un comportamiento localmente invasivo.
Representa del 1 – 3 % de los tumores cerebrales primarios y se manifiesta con una Incidencia bimodal con picos entre los 5 - 14 y 55 – 65 años.
Se presenta en 3 variantes histológicas:
• Quiste epitelial mucoide : con células secretoras de mucina.
• Variante adamantinomatosa : contiene áreas de epitelio escamoso y calcificaciones. Es la forma más frecuente hallada en niños y adolescentes.
• Variante escamosa – papilar : se compone únicamente de epitelio escamoso y no presenta calcificaciones. Es la forma más frecuentemente hallada en adultos.
Los síntomas dependen del compromiso de estructuras vecinas:
• Síntomas de hipertensión endocraneal : cefalea, vómitos.
• Alteraciones visuale s: disminución de la agudeza y defectos campimétricos.
• Alteraciones endocrinológicas (80%) : déficit de GSH que se expresa como impotencia sexual o amenorrea (90%), hiperprolactinemia (50%), hipotiroidismo (40%), alteración de la hormona del crecimiento (40%), insuficiencia suprarrenal (25%) y diabetes insípida (10%).
Para el diagnóstico el método de elección es la RMN que permite establecer la localización y extensión del tumor, además del estudio anatomopatológico para su confirmación.
Para su tratamiento puede optarse por una resección completa o parcial asociada a radioterapia. La primera de ellas suele evolucionar con un mayor grado de morbimortalidad y secuelas neurológicas pero con una menor tasa de recidivas (10%) a diferencia de la segunda que tiende a optarse en tumores de mayor tamaño, con importante componente quístico y calcificaciones. La vía de abordaje puede ser transcraneal o transesfenoidal con una mortalidad global del 2-3% y una tasa global de supervivencia a los 10 años del 90%.
Respecto a los trastornos pigmentarios cutáneos, recordemos en primer lugar que la coloración de piel está determinada por la presencia de melanina, la vascularización de la misma y los carotenos. De ellos adquiere un papel fundamental la melanina la cual es secretada por los melanocitos a partir del estímulo que se genera por la melanotrofina (MSH) producida por la hipófisis en relación a la presencia de factores liberadores o inhibidores de la misma desde el hipotálamo. Otra hormona capaz de generar el mismo estímulo es la adrenocorticotrofina (ACTH) y esto se debe a que ambas comparten características estructurales por provenir del mismo precursor: la propiomelanocortina. Los trastornos pigmentarios cutáneos van desarrollarse tras alteraciones a nivel de la producción, distribución o metabolismo de la melanina. Estos consisten en hipercromía, hipocromía o acromía. De ellos me voy a referir a los casos de hipo y acromía.
Como se observa en la siguiente tabla, estas enfermedades cutáneas pueden ser congénitas o adquiridas.

Dado que nuestro paciente no presentaba ninguna dermatosis inflamatoria o infecciosa previo al inicio de este cuadro y negaba contacto directo con sustancias químicas, voy a referirme al vitíligo como trastorno pigmentario adquirido capaz de producir lesiones de este tipo. Vitíligo .
Se presenta en el 1 – 2% de la población mundial afectando tanto la piel y el pelo como así también células pigmentadas del globo ocular y del oído interno.El 50% de los pacientes desarrolla la enfermedad antes de los 20 años y el 70% lo hace antes de los 30.
En el desarrollo del vitíligo existe un componente genético asociado a factores medioambientales y emocionales que pueden actuar como precipitantes.
Las manifestaciones clínicas se caracterizan por la presencia de máculas acrómicas, de variable tamaño y confluyentes que se localizan preferentemente en cara, cuello, dorso de manos y sitios periorificiales. Puede adquirir también una distribución metamérica.
La distribución puede ser localizada o generalizada. En el primer grupo se describen lesiones focales y segmentarias, mientras que en el segundo adquieren una localización vulgar, acrofacial o universal. Estas pueden observarse en el siguiente gráfico.
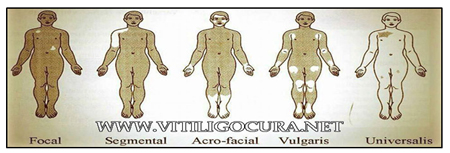
Respecto a la patogenia, aún no ha sido establecido el mecanismo que produce la presencia de estas lesiones aunque se han propuesto hipótesis respecto a la misma. • Hipótesis Inmunitaria: se produciría una reacción anti-melanocitos en relación a la presencia de anticuerpos anti receptores de superficie mediada por linfocitos T CD8. Los fundamentos que avalarían esta teoría es la relación existente entre vitíligo y otras enfermedades autoinmunes, como ser: tiroiditis, diabetes, enfermedad de Addison y anemia perniciosa y la mejoría que se evidencia en las lesiones tras los tratamientos con fármacos inmunomoduladores.
• Hipótesis neurocitotóxica : existirían sustancias de las terminaciones nerviosas (neuropéptidos o catecolaminas) con efecto tóxico sobre el melanocito y el metabolismo de la melanina. Esta teoría se sustenta en la distribución metamérica que pueden adquirir las lesiones, en la presencia de enfermedades neurocutáneas (neurofibromatosis, esclerosis tuberosa) y en la relación evidenciada entre la presencia de vitíligo y estrés emocional.
• Defecto intrínseco del melanocito: aún se encuentra en estudio pero plantea un posible defecto en el retículo endoplásmico del melanocito que determina una temprana apoptosis .
Para el tratamiento del vitíligo se debe tener en cuenta en primer lugar que, si bien el curso es impredecible, el 73% presenta progresión de la enfermedad. Tanto la leucotriquia como la presencia de un fenómeno de Koebner son factores de mal pronóstico. El tratamiento puede ser tópico cuando la lesión ocupa menos del 20% o sistémico si afecta entre el 20 y 50% de la superficie corporal. Cuando la afección supera el 50% puede optarse por la despigmentación de piel sana. Para el mismo se utilizan sustancias con efecto fotoestimulante e inmunomodulador. Entre los primeros se encuentran los psolarenos (8-metoxipsolareno, trimetilpsolareno) que requieren de la posterior exposición solar y en el segundo grupo hallamos a los corticoides y los inhibidores de la calcineurina (tacrolimus) que pueden ser administrados de forma tópica o sistémica. El tratamiento quirúrgico nunca es de primera elección y se indica en casos de vitíligo localizado y no progresivo. Existen diversas técnicas como son los mini-injertos, la suspensión epidérmica de melanocitos, el injerto epidérmico por succión y los cultivos de melanocitos in vitro. Se debe tener en cuenta además los aspectos psicosociales del vitíligo por ser una enfermedad que modifica negativamente el aspecto estético del paciente.
Respecto a la posibilidad de que en este caso las lesiones se encuentren formando parte de un síndrome paraneoplásico, existe evidencia que pacientes con melanoma desarrollan vitíligo por la presencia aumentada de anticuerpos anti-melanocitos. Por otro lado se han descripto escasos reportes de casos donde se ha hallado un componente melanocítico en la anatomía patológica de los craneofaringiomas. Por tales motivos, si debo establecer un solo factor que justifique todo los síntomas que presenta nuestro paciente, considero que podríamos encontrarnos ante un caso de craneofaringiomamelánico, por lo que el estudio anatomo-patológico del mismo resultará fundamental. De no ser así, podríamos inferir algún trastorno a nivel de eje hipotálamo – hipofisario en relación al tumor supraselar o que en su desarrollo intervengan también otros factores medioambientales y emocionales que generen lesiones hipocrómicas.
Bibliografía
• González, S. “Vitiligo”. Carrera de posgrado de Dermatología. Hospital Provincial de Rosario. 2006.
• Páramo, C.; Picó, A.; “ Guía clínica del diagnóstico y tratamiento del craneofaringioma y otras lesiones paraselares”. Grupo de Trabajo de Neuroendocrinología de la Sociedad Española de Endocrinología y Nutrición. 2007.
• Wing, A.;, Charmaine H.; “Vitiligo as a Paraneoplastic Syndrome Preceding Pituitary Adenoma and Subsequent Acute Lymphoblastic Leukemia”. 2015.
• Harris, BT.; Horoupian DS. “Melanoticcraniopharyngioma: a report of two cases”.En Pubmed.gov. 1999.
• Nandita G.; Ravi, D. “ Melanoticcraniopharyngioma, an unusual variant in a young female”. Publicación de la SociedadNeurológica de India. 2010.
|
