Discusión del caso clínico. | Presentación |
Se discutirá el caso de un paciente varón de 32 años, con síndrome febril y anemia hemolítica.
Como objetivos de este seminario propongo:
-
Repasar el abordaje y clasificaciones de anemias hemolíticas.
-
Considerar la utilidad del test de coombs.
-
Analizar posibles causas de fiebre y esplenomegalia en nuestro paciente.
-
Realizar una conclusión.
El caso clínico se trata de un paciente oriundo de Paraguay, que vive desde hace dos años en la ciudad de Rosario, y que realizó recientemente (un mes) un viaje a su ciudad natal, que consulta por un cuadro de 48 hs de evolución caracterizado por fiebre y debilidad generalizada. A su ingreso se realiza un examen físico exhaustivo en el que se constata la presencia de fiebre, se detectan las escleras sub ictéricas y esplenomegalia; y se realiza un análisis de sangre en el que se evidencia anemia con reticulocitosis, hiper bilirrubinemia, y elevación de enzima láctico dehidrogenasa (LDH). Se complementa el estudio del paciente con un análisis de orina que no presenta alteraciones y un frotis de sangre periférica (FSP) en el que se detecta la presencia de esferocitos y 6% de células irritativas. Se realiza también un test de coombs que resulta negativo y un segundo test de coombs que también resulta negativo.
Durante la internación se instaura tratamiento sintomático (antitérmicos) y se evidencia ascenso progresivo de glóbulos rojos con mejoría progresiva de la anemia, y descenso de valores de bilirrubina y LDH, de carácter espontáneo. El paciente no repite registros febriles, y persiste la esplenomegalia. Se realiza una tomografía de tórax, abdomen y pelvis que solo evidencia la presencia de adenopatías inguinales y axilares pequeñas, sugestivas en primera instancia de proceso infeccioso.
Analizando los datos antes mencionados podemos conversar inicialmente sobre la presencia de anemia (que según la OMS, la anemia es un trastorno en el cual el número de eritrocitos resulta insuficiente para satisfacer las necesidades del organismo) y en este caso la anemia es de carácter normocítica y normocrómica, con reticulocitosis, lo que significa que el paciente presenta una medula ósea funcionante.
Siguiendo el protocolo de estudio de las anemias según la Sociedad argentina de Hematología podemos considerar en primera instancia las anemias con volumen corpuscular normal y entre las causas más frecuentes citamos las “anemias post hemorrágicas” que se presentan fundamentalmente luego de traumatismos, o sangrados digestivos entre otras causas, que descartamos tras realizar un interrogatorio y examen físico minucioso o por ejemplo con la búsqueda de sangre oculta en materia fecal. Posteriormente vamos a considerar las “anemias hemolíticas” que se caracterizan por aumento en la destrucción de los eritrocitos que supera la capacidad de regeneración medular.
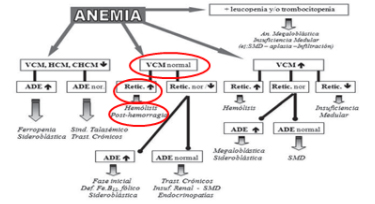
Una vez dentro de las clasificaciones de anemias hemolíticas, podemos dividirlas según el sitio donde se produce la hemolisis, en intravasculares y extravasculares. En las primeras la hemolisis ocurre en el interior de los vasos sanguíneos y produce liberación de la hemoglobina hacia el torrente circulatorio, donde se une a la haptoglobina ocasionando un descenso en su nivel de concentración, y causando además la presencia de hemoglobinuria y hemosiderinuria en un examen de orina, que de manera comparativa con nuestro paciente, en el análisis de la orina no había evidencia de hemoglobinuria.
Cuando la destrucción es de localización extravascular, se produce fundamentalmente en el bazo sanguíneo, donde se separan todos los componentes eritrocitarios, por un lado el hierro que se acumula en forma de depósitos hepáticos, por otro lado las globinas que se reutilizan y por último la protoporfirina libre, que se transforma en biliverdina y luego en bilirrubina indirecta que atraviesa las membranas plasmáticas y se manifiesta como ictericia cutáneo mucosa y en escleras. Parte de la bilirrubina indirecta se conjuga, transformándose en bilirrubina directa que se libera al intestino, sitio en el cual participa de la digestión y a este nivel las bacterias entéricas la convierten en estercobilinógeno y urobilinógeno, que se filtra a nivel renal ocasionando orinas oscuras. Considero por las manifestaciones clínicas que nuestro caso se trata de una anemia hemolítica de localización extravascular.
Otra de las clasificaciones de anemias hemolíticas consiste en dividirlas según el sitio donde se localiza el defecto que ocasiona la destrucción rápida de los eritrocitos, dentro de esta clasificación podemos citar, las anemias intracorpusculares cuando el defecto se encuentra localizado en la membrana eritrocitaria, generalmente son congénitas; y extracorpusculares cuando el defecto en el eritrocito es secundario a trastornos del medio que los rodea, plasmático o vascular, y generalmente son de causas adquiridas.
Entre las anemias intracorpusculares podemos nombrar en primera instancia a las eritroenzimopatías, que se caracterizan por presentar un defecto en la síntesis o configuración de las enzimas que participan en el metabolismo eritrocitario. Las dos más frecuentes son: Déficit de glucosa-6-fosfato-deshidrogenasa y Déficit de piruvato cinasa. La hemolisis por este mecanismo se desencadena luego de la ingesta de ciertos fármacos (por ejemplo: Antipiréticos, sulfonamidas, antipalúdicos) y consumo de antioxidantes (como por ejemplo: vitamina C), y entre los hallazgos sugerentes se evidencia la presencia de eritrocitos crenados en un frotis de sangre periférica.
En nuestro caso, el paciente no presenta antecedente de consumo de fármacos ni agentes oxidantes previo a la consulta, ni tampoco presenta eritrocitos crenados en FSP, motivo por el cual no considero éste como un diagnóstico.
Luego podemos citar las hemoglobinopatías, que a su vez se subdividen en Cuantitativas o estructurales y cualitativas o talasémicas. Entre las primeras existen al menos 400 tipos, y se caracterizan por trastornos en la codificación de la cadena de aminoácidos constituyentes de las cadenas de la molécula de hemoglobina, de los cuales el tipo más frecuente es la hemoglobinopatía de células “s” (anemia falciforme). Las hemoglobinopatías cualitativas se caracterizan por déficit total o parcial de cadena beta o alfa de la hemoblogina. Este tipo de anemia se caracteriza por la presencia de drepanocitos en FSP, y son además anemias de tipo microcíticas e hipocrómicas, por estos motivos creo que nuestro paciente no presenta una hemoglobinopatía como causa de su trastorno hematológico.
Finalmente en el último grupo de las anemias corpusculares se encuentran las membranopatías, que se dividen en dos sub tipos, la Eliptocitosis hereditaria que es menos frecuente dado que generalmente es asintomática por lo que pasa desapercibida y no llega a diagnosticarse, y la Esferocitosis hereditaria que se caracteriza fundamentalmente por la presencia de esferocitos en FSP. Dado que nuestro paciente presenta dichas células, considero interesante hacer una breve extensión en cuanto a este trastorno ya que lo considero como un probable diagnóstico.
La “Esferocitosis hereditaria” es una membranopatía considerada como la anemia hemolítica congénita más frecuente, y si bien su diagnóstico casi siempre se realiza en la infancia, también puede diagnosticarse en etapa adulta, (dato documentado en revisión de Esferocitos hereditaria, de la sociedad argentina de hematología). Se lo considera un trastorno de origen congénito, presenta un 75% de transmisión genética de carácter autosómico dominante donde uno de los dos progenitores presenta síntomas clínicos o hallazgos de laboratorio compatibles, y el 25% restante es de transmisión autosómica recesiva donde ninguno de los progenitores presenta ni clínica ni hallazgos de laboratorio. En cuanto a la patogenia, el defecto está determinado por una alteración en la configuración de la membrana plasmática de los eritrocitos (dicha membrana está formada por lípidos y proteínas, y este trastorno se presenta con mayor frecuencia por alteraciones en la configuración a nivel proteico), y en consecuencia se pierde parte de la superficie eritrocitaria que ocasiona mayor fragilidad osmótica y causa distorsión de la forma habitual del glóbulo rojo, que adquiere forma de “globo” y al atravesar los sinusoides en el bazo quedan retenidos y finalmente lisados.
Con respecto a la clínica: varía desde portador asintomático a estadíos de hemolisis que pueden ser leves, moderados o severos en base al valor de hemoglobina y al requerimiento de transfusiones ante las crisis hemolíticas, las que generalmente se desencadenan de manera secundaria a procesos infecciosos virales. Cabe destacar que en casi todos los casos sintomáticos los pacientes presentan anemia, ictericia y esplenomegalia de carácter progresivo.
Por último el diagnostico se realiza con el FSP que evidencia los “esferocitos” que son las típicas células del trastorno y que también pueden someterse al test de la fragilidad capilar. Respecto al tratamiento consiste en terapia de sostén transfusional frente a las crisis hemolíticas y en los casos severos que requieren transfusiones sanguíneas diarias se plantea la realización de esplenectomía.
Dentro de las causas de anemias de origen corpuscular, considero que nuestro caso podría tratarse de Esferocitosis hereditaria, que se presenta en esta oportunidad en forma de crisis hemolítica, y si al estudiar familiares del paciente no encontramos evidencia clínica ni analítica en sus progenitores, podemos inferir también que el paciente presenta un patrón de herencia de tipo autosómico recesivo.
Para considerar dentro de las anemias hemolíticas de origen extracorpusculares, podemos citar las anemias Aloinmunológicas que se presentan luego de una transfusión sanguínea, debido a que en nuestro caso el paciente niega este antecedente y no recibió transfusiones durante la internación, no lo consideraré en esta discusión.
Otra entidad es la Hemoglobinuria paroxística nocturna que también descarto dado que es un tipo de anemia que se caracteriza además por afectación de las tres series medulares, y respecto a la clínica presenta manifestaciones propias de fenómenos trombóticos o aplásicos. Creo que se encuentra alejado de ser un probable diagnostico en este caso.
Finalmente, otorgando relevancia al test de Coombs (prueba útil ante anemia hemolítica) como bien sabemos las anemias de causa autoinmune se clasifican en: anemias hemolíticas por anticuerpos calientes (Ig G), que siempre cursan con test de coombs positivo; y anemias hemolíticas por anticuerpos fríos (Ig M o crioaglutininas) que generalmente son de causa secundaria y pueden presentarse con test de coombs negativo, ya que la lisis eritrocitaria finalmente se produce a través de la fracción Fc del complemento y al realizar test de coombs utilizando por ejemplo suero mono específico para Ig G, el resultado final es negativo.
Otra causa de test de coombs negativo son las anemias hemolíticas inmunomediadas por Ig A, que según un artículo publicado en American Jornal of Hematology, en ellas se produce depósito de Ig A en la membrana eritrocitaria y se cree que de esta forma, el glóbulo rojo tras sensibilizarse por Ig A es capturado y secuestrado en el bazo, ocasionando su destrucción y consecuente anemia hemolítica sin activación del sistema inmunológico. Es una de las causas más raras de anemias hemolíticas con test de coombs negativo que involucra anticuerpos. Es un trastorno raro con una frecuencia muy baja.

Luego de esta breve revisión y tras consultar sobre los test de coombs que se realizan en nuestra institución (Hospital Provincial del Centenario), sabiendo que incluyen suero de coombs Ig G y fracción C3d del complemento creo poco probable que se trate de un proceso de origen autoinmune por anticuerpos calientes, tampoco considero que se trate de un proceso inmunomediado por Ig A, dado que son casos raros con muy baja frecuencia. Y luego de repasar causas secundarias de anemia hemolítica por anticuerpos fríos tampoco lo considero un diagnostico probable ya que nuestro paciente no presenta síntomas respiratorios y se la realizó una tomografía de tórax que no evidenció hallazgos patológicos.
Ahora bien, nuestro paciente presenta fiebre y esplenomegalia, y me permito preguntarme: ¿tiene un proceso infeccioso?
Para repasar algunos conceptos básicos, recordemos que el bazo se encuentra en el hipocondrio izquierdo, en íntimo contacto con la curvatura gástrica, la cola del páncreas, el riñón izquierdo y el colon. Tiene un peso aproximado de 150 g, y mide 12 x 7 cm.
Hablamos de Esplenomegalia cuando es clínicamente palpable, para lo cual debe aumentar entre 2 y 3 veces su tamaño habitual.
Las esplenomegalias son sintomáticas cuando ocasionan distensión o pesadez abdominal, dolor o son palpables, y pueden acompañarse o no de Hiperesplenismo (aumento en la funcionalidad del bazo).
Existen tres causas importantes responsables de hiperesplenismo: Estasis vascular, por ejemplo secundario a cirrosis e hipertensión portal (nuestro paciente no presenta habito etílico, tiene valor de colinesterasa normal en hepatograma y no se observan indicios de dicho hallazgo en la ecografía realizada). Las neoplasias, sobre todo las hematológicas (el paciente fue sometido a una tomografía en busca de procesos linfoproliferativos sin evidencia) y finalmente las infecciones, en primer lugar las virales.
Analizando nuestro paciente febril, oriundo de Paraguay y que realizó un viaje a su ciudad natal hace poco más de un mes, por considerarlo noción de foco se merece una mención aparte el Paludismo, que es una enfermedad endémica ocasionada por parásitos protozoarios. Cuyo agente es el Plasmodium y su vector el mosquito hembra del genero anopheles. Su periodo de incubación va desde 8 a 40 días. El humano es el huésped definitivo donde se completa el ciclo asexual de la vida del parásito. Consta de dos fases, una hepática y una eritrocitaria y ocasiona anemia hemolítica de tipo corpuscular intravascular. Respecto a la clínica el síntoma cardinal es la fiebre, llamada “fiebre cuartana” diaria, de carácter cíclico, que consta de tres fases bien definidas: la primera: escalofríos, seguido de periodo de 2-4 hs de duración de fiebre de 39-40° y finalmente periodo de sudoración donde se produce un descenso brusco de la temperatura.
En cuanto a los métodos diagnósticos el Gold Estándar es la Gota gruesa (microscopía de luz visible) que debe realizarse de manera seriada cada 6 hs durante 48 hs, aunque se considera positivo si la primera muestra arroja resultado positivo. Se acompaña además de alteraciones en el hepatograma e hiperbilirrubinemia marcada.
Considero en nuestro caso, alejada la posibilidad de tratarse de un paludismo, porque si bien estamos dentro del período de incubación, el paciente no presenta clínica ni alteraciones analíticas compatibles con esta enfermedad.
Para acercarnos al final, resta mencionar entre las causas de fiebre y esplenomegalia, los procesos infecciosos virales. Recordemos también que el FSP del paciente en cuestión presenta 6% de células irritativas (linfomonocitarias) que normalmente están ausentes cuando no existe infección activa.
Por considerar un porcentaje no despreciable y por tanto, debemos otorgarle relevancia infiriendo que en este momento nuestro paciente presenta un proceso infeccioso de etiología viral. Y considerando también la esplenomegalia, fiebre, y adenopatías (evidenciadas en tomografía), podríamos englobar dichos hallazgos dentro de un probable síndrome mononucleosiforme y en orden de jerarquía las causas más frecuentes son: Virus de Epstein Barr (VEB), Citomegalovirus (CMV) y finalmente Parvovirus B19, los tres virus pertenecientes a la familia Herpesviridae, cuya patogenia además ser inmunomediada también ejercen citotoxidad directa. Algunas fuentes bibliográficas consideran al Virus Herpes Simple (VHS) entre las causas de síndrome mononucleosiforme aunque sus principales manifestaciones sean ulceras genitales o bucales. Tras repasar la clínica habitual de este último y luego de revisar a nuestro paciente de manera exahustiva no considero que estemos frente a una infección por VHS.
Respecto a la infección por parvovirus B19, su forma de presentación más frecuente es la aplasia de la serie roja dado que particularmente ejerce toxicidad celular intramedular directa.
En nuestro caso no hay evidencia de aplasia roja sino una probable crisis hemolítica y por este motivo creo que queda algo alejado como primer diagnóstico.
Luego considerando infección por Citomegalovirus; se encuentra bien documentado que es más frecuente en pacientes con trastornos de la inmunidad y pacientes trasplantados (que no es nuestro caso), de cualquier manera luego de una intensa búsqueda bibliográfica existe evidencia de casos que comentaré a continuación.
Y por último considerando la infección por Virus de Epstein Barr sus manifestaciones se ajustan a nuestro caso problema tanto por manifestaciones clínicas como en exámenes complementarios (crisis hemolítica, células irritativas, fiebre, esplenomegalia, adenopatías) por tal motivo lo considero dentro un probable diagnóstico.
Para ilustrar mi discusión y analizando la bibliografía considero interesante incluir algunos reportes de casos. En primera instancia incorporo un reporte y revisión sobre un caso de “anemia hemolítica por citomegalovirus en adulto inmunocompetente”. Esta revisión describe el caso clínico de un adulto varón de 44 años, en el que se documentó la presencia de anemia hemolítica con test de coombs negativo (similar a nuestro caso). Y a raíz del hecho se realizó una revisión de literatura entre los años 1980 y 2008 en la que se incluyeron 290 adultos inmunocompetentes con manifestaciones severas debidas a infección por Citomegalovirus y en 12 de ellos se documentó la presencia de anemia hemolítica, de los cuales 3 presentaron test de coombs negativos.
Este reporte presenta similitud con nuestro paciente dado que se trata de un adulto varón inmunocompetente que también presenta anemia hemolítica hemolítica con test de coombs negativo.
Incluyo también otro reporte de caso del año 2012, que describe un caso de “anemia hemolítica como manifestación inicial de esferocitosis hereditaria inducida por infección por parvovirus B19 y Virus Herpes simple, en paciente con Talasemia”, si bien podría adaptarse a nuestro paciente respecto a la primera manifestación inicial de trastorno hereditario diagnosticado en edad adulta secundario a proceso infeccioso viral, discrepa en cuanto al antecedente de diagnóstico previo de talasemia, de cualquier manera es un caso interesante para ilustrar nuestra discusión.
Y por último incluyo un reporte de caso de aplasia de la serie roja secundario a infección por parvovirus B19 en un paciente con esferocitosis hereditaria. Dicho caso presenta como similitud a nuestro paciente la evidencia de la hemolisis con test de coombs negativo, y la presencia de esferocitos en frotis de sangre periférica, pero difiere en que el caso documentado es un paciente de 4 años y nuestro caso se trata de un adulto mayor de 30 años.
No encontré clara evidencia de hemolisis por virus de Epstein barr pero considero que al tratarse de un virus de igual familia que el resto de virus documentados y la forma de presentación de nuestro paciente se ajusta en varios aspectos y podría considerarse un probable diagnóstico.
Luego de todo lo descripto y para ir concluyendo considero que; en este paciente varón de 32 años inmunocompetente, con anemia hemolítica, esplenomegalia y esferocitos en sangre periférica que además presenta fiebre y hallazgo de células irritativas en frotis de sangre periférica, podríamos estar frente a la primera manifestación de Esferocitosis hereditaria inducida por infección viral (Epstein Barr o Parvovirus B19, como etiologías más probables).
Es por esto que propongo: esperar resultados de serologías virales a fin de documentar la infección; control evolutivo y continuar estudio del paciente de manera ambulatoria, explorando además causas de anemias post hemorrágicas; y estudiar a su familia en busca de esferocitosis teniendo en cuenta además que puede estar ausente y tratarse de herencia autosómica recesiva.
BIBLIOGRAFÍA:
-
M.J. García Rodrígueza, Protocolo diagnóstico de las anemias hemolítica. RevMedicine. 2008; 10 (20) 1371-4
-
Aixalá, Mónica, Basack, Nora Eritropatías. Sociedad Argentina de Hematología • GUIAS DE DIAGNOSTICO Y TRATAMIENTO • 2015 Tomo 1. Pag7-130
-
E. Feliu Frasnedo, Principios generales de la exploración del enfermo con hemopatía. Farreras-Rozman XVI edición, volumen II, Sección 14-203. Pag 1627-1652.
-
M. T. Hernandez García, Enfermedades del sistema eritrocitario.Farreras-Rozman XVI edición, volumen II, Sección 14-204. Pag 1654-1678.
-
J. Vaqué Rafart, Epidemiología general de las enfermedades trasmisibles. Farreras-Rozman XVI edición, volumen II, Sección 17 (parte 1)-250. Pag 2202-2206.
-
P. Coll Figa, Diagnóstico de enfermedades infecciosas. Farreras-Rozman XVI edición, volumen II, Sección 17 (parte 1)-251. Pag 2206-2213.
-
C. Muñoz Batet, Infecciones causadas por protozoos apicomplexa hemotisulares. Farreras-Rozman, XVI edición, volumen II, Seccion 17 (parte 4)-298. Pag 2447-2456.
-
J. L. Perez, Infecciones causadas por citomegalovirus. Farreras-Rozman, XVI edición, volumen II, sección 17 (parte 5)-307. Pag 2497- 2501.
-
M. Gurgui Ferrer, Infecciones causadas por el virus de Epstein-Barr. Mononucleosis infecciosa. Farreras-Rozman, XVI edición, volumen II, sección 17 (parte 5)- 308. Pag 2501-2505.
-
Jesus F San Miguel, Hematología. Manual básico razonado. 3° edición. Elsiever.
-
J. L. Rodriguez García, GreenBook edición 2013. ISBN: 978-84-7101-924-0. Sección hematología. Adenopatías S 76 Pag 891-908; Esplenomegalia S85 Pag 1014-1017.
-
Nicole D. Zanteck, The direct antiglobulin test: A critical step in the evaluation of hemolysis. Am. J. Hematol. 87:707–709, 2012.
-
Philippe Chadebech, IgA-mediated human autoimmune hemolytic anemia as a result of hemagglutination in the spleen, but independent of complement activation and Fc_RI. INMUNOBIOLOGY. Journal 2010;116(20):4141-4147).
-
Segel GB, Direct antiglobulin ("Coombs") test-negative autoimmune hemolytic anemia: a review. Department of medicine, NY USA. PubMed 2014, January 9. 52(4):152-60.
-
J. Med case report: Hemolytic anemia due to acute cytomegalovirus infection in an immunocompetent adult: a case report and review of the literature. Published online 2010 Oct 21. doi: 10.1186/1752-1947-4-334.
-
Veldhuis W. Abstract: Coombs-negative severe haemolytic anaemia in an immunocompetent adult following cytomegalovirus infection. Eur J Clin Microbiol Infect Dis. 2004 Nov;23(11):844-7. Epub 2004 Oct 20.
-
Vivens Corrons JL. Anemias hemolíticas: aspectos generales. En: Sans-Sabrafen J, Besses Raebbel C, Vives Corrons JL, editores. Hematología clínica. 5ª ed. Madrid: Elsevier; 2006. p. 187.2. Gehrs BC, Friedberg RC. Autoimmune hemolytic anaemia. Am J Hematol. 2002;69:258-71.
-
Sigbojrn Berentsen, Diagnosis and treatment of cold agglutinin mediated autoimmune hemolytic anemia. Blood Reviews 26 (2012) 107–115.
|
Imágenes del caso
|
