Discusión del caso clínico. | Presentación |
Se discutirá el caso de una mujer de 55 años con síndrome cerebeloso y retención aguda de orina.
En primera instancia me planteo los siguientes objetivos:
-
ANALIZAR CAUSAS DE SINDROME CEREBELOSO.
-
PLANTEAR PROBABLES DIAGNÓSTICOS DIFERENCIALES.
-
NOMBRAR CAUSAS DE RETENCIÓN AGUDA DE ORINA.
-
PROPONER ALGUNOS PASOS A SEGUIR.
Para comenzar esta interesante discusión considero oportuno analizar la signosintomatología de nuestra paciente dado que si bien inicialmente todo apuntaba a tratarse de un síndrome vertiginoso de origen periférico, posteriormente encontramos datos en exámenes complementarios, examen físico y antecedentes de la paciente que nos llevaron a buscar un poco más lejos y es por eso que podríamos englobar todos los hallazgos dentro de lo que comúnmente conocemos como Síndrome Cerebeloso.
El cerebelo que ocupa la mayor parte de la fosa posterior, es el encargado de regular las informaciones sensitivas aferentes y coordinarlas con los estímulos eferentes motores procedentes del cerebro, permitiendo la realización de movimientos finos y de precisión. Junto a esta coordinación de los movimientos, regula y controla el tono muscular. Cuando se produce alguna alteración cerebelosa se producen alteraciones de la estática y la marcha, de las adaptaciones posturales y también del gesto. Dentro de los hallazgos clínicos podemos citar: dismetría, adiadococinesia, hipotonía, discronometría, disartria, nauseas, mareos y alteraciones en la marcha. Estos últimos presentes en nuestra paciente.
Cuando hay un trastorno del cerebelo los mecanismos reguladores se alteran, dando lugar por ejemplo a la ataxia cerebelosa, que es debida fundamentalmente a la pérdida de armonía entre los músculos agonistas y antagonistas y se caracteriza por aumento de la base de sustentación, con brazos en abducción y desvío lateral (Romberg +, lateropulsión, aumento de la base de sustentación, nauseas, nistagmo) Todos estos signos están presentes en nuestra paciente.
Algoritmo de estudio inicial ante un paciente con síndrome cerebeloso:
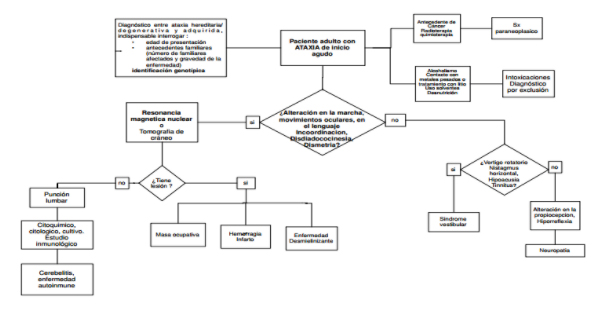
Una vez analizado dicho algoritmo vamos a definir algunas cuestiones.
Ante cuadros neurológicos agudos y según la clínica que el paciente presente, es de utilidad la realización de punción lumbar. En nuestro caso se realizaron dos punciones lumbares separadas por cinco días de diferencia. En la 1° se obtuvo un recuento de 44 elementos/mm3 y en la 2° un recuento de 26 elementos/mm3. Descenso interpretado como de carácter espontáneo dado que no hubo mediación, hasta entonces, de tratamiento alguno.
Y en este caso la pregunta sería: ¿Estamos frente a una meningitis?
Se considera Líquido cefalorraquídeo normal (LCR): cuando presenta presión de apertura de 8-12 cmH20, aspecto claro, celularidad < 5 cel/mm3, proteínas < 50mg%, Glucosa > 40 mg/dl o relación LCR/plasma > 0,40.
Hallazgos sugestivos de meningitis:
-
Tinción de Gram positiva.
-
Glucorraquia < 40 mg/dl, o relación glucosaLCR/glucosaPlasma < 0,40.
-
Proteinorraquia > 200 mg/dl.
-
Glóbulos blancos > 1000 mc/microL.
-
Recuento diferencial de polimorfonucleares > 80%.
-
Presión de apertura > 20 cmH20
En este caso no presenta hallazgos sugestivos de meningitis, no considero que se trate de un proceso de etiología viral/bacteriana. Pero ante la evidencia de leve aumento en el número de elementos con mejoría progresiva, podemos considerar que se trata de un líquido claro de tipo inflamatorio.
Como etiología compatible con este resultado de LCR citamos los procesos infecciosos autoinmune, en particular la “Encefalitis autoinmune”, entidad que en los últimos años ha aumentado el número de reportes de casos, y se asocia con anticuerpos dirigidos contra las superficies neuronales o proteínas sinápticas generando manifestaciones neurológicas pero sin fiebre y LCR con pleocitosis (como en nuestro caso). Según artículo publicado en IntraMed existen criterios diagnósticos específicos y se sugiere examen neurológico exhaustivo, y complementar con imágenes por ejemplo, Resonancia Magnética (RMI), Electromiografía (EMG).
En nuestro caso se cumple por el momento la pleocitosis > 5 cel/mm3, pero ninguno de los criterios propuestos. Por tal motivo no lo considero probable diagnóstico.

Otras causas de síndrome cerebeloso, son las masas ocupantes de espacio no infecciosas, entre las que podemos considerar los tumores intracraneales y dentro de ellos citar Neoplasias 1° (supratentoriales e infratentoriales), Neoplasias metastásicas generalmente multifocales (los tumores que metastatizan en SNC con mayor frecuencia son: cáncer de pulmón, mama y melanoma maligno), y también los síndromes paraneoplásicos.
La clínica de las neoplasias 1° o 2° se clasifica en focal y general según el sitio donde se localice la masa tumoral, y según produzcan o no síntomas de hipertensión intracraneal.
Según la bibliografía consultada, en adultos son más frecuentes: glioblastoma multiforme, astrocitoma anaplásico, seguidos de meningiomas, tumores de la hipófisis y neurinomas, estos últimos de predominio en mujeres, y los linfomas en pacientes con compromiso de la inmunidad (HIV, tratamiento inmunosupresor, transplantados).
Por la localización de afectación en las imágenes iniciales de cráneo de nuestra paciente se consideró como probable diagnostico el Linfoma primario del sistema nervioso central (LPSNC), que es un tipo de linfoma No Hodking de células B, de carácter agresivo, etiopatogenia desconocida, representa aproximadamente el 5% de las neoplasias primarias intracraneales. Su incidencia se encuentra en relación al estado de inmunosupresión y puede asociarse a infección por Virus de Epstein Barr (VEB).
En pacientes inmunocompetentes, el LPSNC suele presentarse con una lesión única de localización hemisférica central, que afecta la región supratentorial y a menudo alcanza o cruza la línea media a través del cuerpo calloso, al cual tiende a afectar.
La médula espinal es el sitio más inusual de afectación. Pese a la quimioterapia, las tasas de morbimortalidad son altas debido a las lesiones multifocales del SNC.
El diagnóstico definitivo se establece a través de biopsia estereotáxica, y pueden aparecer células malignas en citometría de LCR hasta en un 40% de los casos. La obtención de citología positiva puede sustituir la realización de biopsia. Suele cursar con hiperproteinorraquia en LCR. Respecto al tratamiento son tumores sensibles a la terapia con corticoides, pudiendo incluso desaparecer durante algunas semanas.
A modo ilustrativo de lo antedicho, cito un artículo publicado en Revista de Medicina de Buenos Aires (MEDICINA, Buenos Aires. Febrero 2017; 77: 17-23), de un estudio retrospectivo de 48 casos de LPSNC diagnosticados en FLENI. Resultados: el 85% de los pacientes incluidos fueron adultos inmunocompetentes. EL 10% de ellos se presentó con clínica compatible con síndrome cerebeloso. Sólo en 11% (2 casos en LCR y 1 en Humor vítreo) la citología fue positiva para células tumorales.
Adjunto debajo RMI con evidencia de compromiso de cuerpo calloso en nuestra paciente, de manera comparativa con imagen obtenida de la serie de casos comentada.
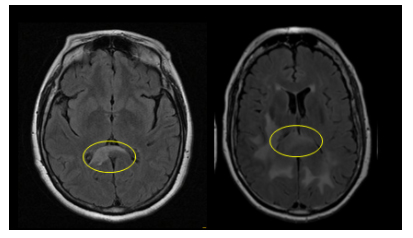
Ahora bien, de manera comparativa, nuestra paciente presenta en RMI lesiones inespecíficas múltiples supra e infratentoriales, que atraviesan línea media en esplenio del cuerpo calloso, que podrían corresponder a un LPSNC, pero también presenta alteraciones inespecíficas en médula sacra (hallazgos inusual en LPSNC), no presenta inmunodeficiencia, lo que constituye un importante factor de riesgo para padecer esta entidad y además frente a la sospecha imagenológica inicial se solicitó la citometría de LCR en la que no se evidencian blastos, y dos muestras de LCR con proteinorraquia normal, que aleja aún más este probable diagnóstico.
Otra entidad que debe sospecharse ante cuadros similares al nuestro, y con evidencia de imágenes inespecíficas en RMI, son los Síndromes Paraneoplásicos, que constituyen síndromes que aparecen en relación a un cáncer generalmente oculto. Son raros, con una incidencia menor al 1%, y en cuanto a las manifestaciones clínicas, son de presentación subaguda. Como probable hipótesis se cree que existen autoanticuerpos antineuronales específicos, la respuesta inmune que inicialmente se dirige contra células tumorales afecta al sistema nervioso que tiene antígenos idénticos o similares, ocasionando su destrucción.
Los dos síndromes paraneoplásicos más frecuentes son: Degeneración cerebelosa: que se define como síndrome pancerebeloso de evolución rápida por destrucción de las células de Purkinje del cerebelo. Y la Encefalomielitis paraneoplasica: que se caracteriza por pérdida neuronal e infiltrado inflamatorio parenquimatoso y perivascular en diferentes localizaciones del SNC. Respecto a la forma de presentación clínica es variable según el territorio afectado: por ejemplo, encefalitis límbica (afectación de la memoria), afectación de pares craneales o tronco, neuropatías sensitivas.
Esta entidad requiere un alto índice de sospecha, y contribuye a la búsqueda del cáncer, oculto hasta entonces, y el cuadro es irreversible.
Considerando las entidades más frecuentes de síndrome paraneoplásicos, no considero que este sea un caso probable dado que en el curso evolutivo de la internación la paciente presentó mejoría clínica espontánea.
En cuanto al análisis de la etiología vascular como responsable del cuadro, hemorrágica o isquémica, en nuestra paciente la tomografía de cráneo realizada a su ingreso y la angio-RMI posteriormente, no evidencian hallazgos patológicos compatibles con estas entidades, no lo considero probables diagnósticos en esta oportunidad.
Continuando con los diagnósticos diferenciales, en esta paciente con síndrome cerebeloso de inicio agudo con antecedente de retención aguda de orina y que presenta alteraciones imagenológicas voy a mencionar a las Enfermedades desmielinizantes del SNC, que se caracterizan por ser procesos inflamatorios idiopáticos que destruyen selectivamente la vaina de mielina. Su curso puede ser crónico generalmente recurrente; o agudo. Y según el sitio de localización pueden ser difusas o localizadas.
Dentro de las enfermedades desmielinizantes agudas, de afectación difusa una de las entidades más conocidas es la “Leucoencefalopatía multifocal progresiva” (LEMP), que se caracteriza también por presentar afectación multifocal, de carácter progresivo, destructivo e irreversible. Se encuentra íntimamente asociada al HIV como factor de riesgo. Y en su etiología se menciona la infección por el Virus JC. No considero que sea éste un caso de LEMP, dado que nuestra paciente no presenta inmunodeficiencias y al igual que en la comparación con el LPSNC, su cuadro impresiona ser reversible, al menos momentáneamente, como así lo indica la evolución clínica.
También se encuentra dentro de las Enfermedades desmielinizantes agudas difusas, otra entidad conocida como “Encefalomielitis aguda diseminada” (ADEM) proceso desmielinizante que puede afectar tanto la sustancia blanca como la sustancia gris. Ocurre de forma “post infecciosa” (enf. Exantemáticas, infecciones Víricas inespecíficas de tracto respiratorio o gastro-intestinal, mononucleosis infecciosa, entre otras) o “post vaccinal” (secundario a la aplicación de vacuna contra tos ferina, difteria, rubeola, varicela). En un 20-30% puede no encontrarse dicho antecedente. Es más frecuente en la infancia, con leve predominio en el sexo masculino. Se caracteriza por presentarse de forma aguda, en la mayoría de los casos de carácter agresivo. Las manifestaciones clínicas: síntomas de afectación meníngea, encefálica y medular simultáneamente. (Fiebre, cefalea, rigidez de nuca, convulsiones, foco motor, paresia, parestesias). En cuanto al análisis de LCR: existe discreta pleocitosis linfocitaria, son INFRECUENTES las Bandas Oligoclonales (BO). En el análisis de la RMI los signos típicos son: afectación de ganglios basales y tálamo, con alteraciones multifocales BILATERALES, de tamaño considerable, de tipo algodonoso. Esta entidad presenta mortalidad elevada (10-30%), en descenso con terapia corticoide. Puede incluso llegar a una recuperación completa, en casi la totalidad de los casos.
Respecto a su evolución se recomienda control imagenológico en tiempo prudencial con evidencia de desaparición de las lesiones mencionadas.
En cuanto a nuestra paciente; presenta clínica neurológica, probable antecedente de episodio infeccioso (si consideramos la infección urinaria previa), LCR con discreta pleocitosis, aunque proteinorraquia normal (se desconoce resultado de bandas oligoclonales), RMI con lesiones multifocales aunque inespecíficas y de tamaño muy pequeño comparativamente, con respuesta positiva espontánea inicial y posterior a la administración de corticoides. Por todo lo antedicho, considero ADEM como PROBABLE DIAGNOSTICO.
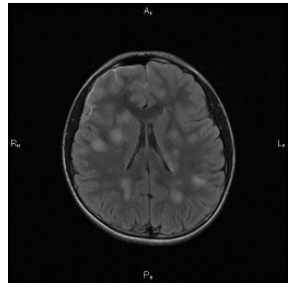
Adjunto aquí una imagen tomográfica de un ADEM típico, con hallazgos patológicos diferentes a los evidenciados en imágenes de nuestra paciente.
Adjunto también un reporte de un caso de una mujer de 50 años que presentó ADEM secundario a una apendicitis perforada, lo que significa que esta entidad puede presentarse de forma secundaria a infecciones más raras.

Y adjunto además otro artículo, reporte de serie de casos muy interesante publicado en la página electrónica de PubMed, en el que se incluyeron 40 pacientes adultos con diagnostico confirmado de ADEM, de ellos 28 fueron mujeres. Y se realizó un seguimiento clínico de 8 a 137 meses. Resultados: 15 pacientes presentaron infección previa. 20% de los pacientes presento LCR normal. Se evidencio la presencia de bandas oligoclonales en el 65%. Casi la totalidad de los pacientes incluidos, (95%) presentó mejoría en fase aguda. Y respecto al seguimiento, en 14 pacientes se realizó diagnóstico de esclerosis múltiple (EM).

Otra entidad desmielinizante de afectación difusa es la “Leucoencefalitis aguda hemorrágica”, que se caracteriza por ser un cuadro hiperagudo y grave, de tipo vasculítico necrotizante, con producción de numerosos focos hemorrágicos difusos, en oportunidades coalescentes en sustancia blanca de hemisferios cerebrales, tronco y medula. En el LCR existe marcada pleocitosis, con hematíes e hiperproteinorraquia marcada. El tratamiento al igual que en ADEM, también se encuentra basado en la administración de corticoides y recambio plasmático.
Considero poco probable que este sea el diagnostico dado que no hay hematíes en LCR, la RMI no evidencia focos hemorrágicos, y en nuestro caso la angio-RMI descarta fenómenos vasculíticos.
Continuando esta revisión sobre las Enfermedades desmielinizantes agudas localizadas se encuentran dos entidades merecen una breve mención: neuritis óptica y mielitis transversa. Inicialmente consideradas como variantes de la Esclerosis Múltiple (EM), pero hoy en día se conocen claras diferencias entre sí. Estas entidades pueden ser agudas o subagudas y asociarse o no, simultáneamente o de manera subsiguiente.
Las manifestaciones clínicas y en RMI difieren de los hallazgos de nuestro caso, y generalmente existen auto anticuerpos en suero Anti-NMO dirigidos contra los canales de aquaporina 4. (No solicitados en nuestro caso por no considerarlo probable diagnostico).
Dentro de las Enfermedades desmielinizantes crónicas, de afectación difusa podemos nombrar la Esclerosis Múltiple (EM): enfermedad de etiología desconocida, con predisposición genética y asociación a factores ambientales, caracterizada por presencia de lesiones focales desmielinizantes en la sustancia blanca denominadas placas, con preservación axonal. Estas lesiones suelen ser múltiples y difusas, y muestran predilección por el tejido periventricular, nervio y quiasma óptico, tronco cerebral, pedúnculos cerebelosos y médula.
Las placas de desmielinización, según la fase de la enfermedad pueden ser agudas (inflamación) o crónicas (desmielinización, degeneración axonal y gliosis). Edad de presentación: en el 70% de los casos entre los 20 y 40 años.
Signosintomatología de inicio: 45% de los casos existe alteración de la sensibilidad, 40% alteraciones motoras, 25% síntomas de disfunción de tronco cerebral (disartria, diplopía, vértigo, nistagmo), 20% afectación cerebelosa (disartria con lenguaje escandido, incoordinación de los miembros e inestabilidad en la marcha). Las alteraciones visuales son infrecuentes como síntomas iniciales, y la afectación esfinteriana es rara como forma de presentación inicial aislada (cuando aparece generalmente se caracteriza por síntomas de incontinencia, irritación vesical, estreñimiento).
Historia natural de la enfermedad: en el 80-90% presentan episodios o brotes de disfunción neurológica, que se repiten en el tiempo y a medida que lo hacen van dejando secuelas funcionales. Curso clínico de la EM: (variable)
-
Recurrente/remitente: brotes con recuperación completa, estabilidad inter episódica.
-
Progresiva primaria: síntomas de inicio gradual con empeoramiento progresivo.
-
Progresiva secundaria: comienza con remisión pero con el tiempo pierde estabilidad inter episódica. Los síntomas continúan empeorando entre las recaídas.
-
Progresiva recurrente: inicio primario progresivo, seguido por recaídas durante el transcurso de la enfermedad.
Hallazgos complementarios: en LCR: hay discreta pleocitosis (<50 cel/mm3), hiper proteinorraquia, y presencia de Bandas Oligoclonales (ausente en suero). RMI: lesiones múltiples desmielinizantes, hiper intensas, difusas en sitios descriptos anteriormente. Potenciales evocados: pueden contribuir al diagnóstico.
El diagnostico se encuentra basado en la concurrencia de la diseminación en el espacio (dos lesiones de diferente localización en SNC), y en el tiempo (dos episodios neurológicos separados en el tiempo). Y además requiere descartar cualquier otra patología que lo justifique.
Para contribuir al diagnóstico de EM, existen los conocidos Criterios de Mc Donald, (revisión 2005): son varios y determinantes: EM definida, probable y NO EM, según si el paciente presenta todos los criterios, algunos o ninguno. En nuestro caso es muy pronto para hablar de EM definida, dado que este cuadro podría sería el primer brote. Deberíamos realizar un seguimiento cercano, a los efectos de poder documentar la aparición de nuevos brotes o nuevas alteraciones diferentes a las actuales en RMI. Por este motivo, recomiendo el estricto control evolutivo una vez externada la paciente.
Y en última instancia no podemos dejar de nombrar el episodio previo de retención aguda de orina. Dentro del capítulo de trastornos vesicales se encuentran las vejigas neurogénicas: que pueden ser de dos tipos: espástica y flácida. En nuestro caso, se trata de una vejiga flácida, en la que no se desencadena el reflejo miccional aunque se distienda la vejiga. Este hecho puede ser provocado por lesiones de las vías sensitivas aferentes o eferentes motoras, o bien lesión del centro medular sacro. Los síntomas de las vejigas flácidas son: retención aguda de orina e incontinencia urinaria. Dentro de las causas más frecuentes se encuentran las litiasis, causas tumorales y también los trastornos desmielinizantes como por ejemplo esclerosis múltiple. Si bien es infrecuente que el primer brote se presente como retención aguda de orina, este antecedente dentro de nuestras sospechas clínicas adquiere relevancia y me permito preguntarme: ¿representa la retención aguda de orina el episodio desencadenante del cuadro neurológico? ¿Es el primer brote de una esclerosis múltiple? ¿Es manifestación de enfermedad desmielinizante?
Ahora bien, luego este análisis del caso clínico, nos encontramos frente a una mujer que presenta un cuadro neurológico con antecedente de episodio reciente de retención aguda de orina, si bien el primer episodio podría haber desencadenado el segundo evento y corresponder a un carácter post infeccioso del cuadro actual, también y más factible creo que podría tratarse de una misma patología que se presenta en dos tiempos, (¿recaída?). En cualquiera de las situaciones, estaríamos frente una manifestación infrecuente de una patología común.
CONSIDERACIONES FINALES: luego de repasar y determinar diagnósticos más probables en esta discusión, propongo:
-
Esperar resultados de bandas oligoclonales y PCR virales solicitadas en líquido cefalorraquídeo.
-
Realizar estudio de potenciales evocados a los efectos de sumar datos a favor de una u otra entidad responsable.
-
Indicar kinesioterapia a los efectos de reducir probables secuelas y/o acelerar remisión sintomática.
-
¿Biopsia estereotáxica? Lo desestimo, dado que es un método complementario cruento, invasivo y en cuanto a riesgo/beneficio no considero apropiada su realización en este momento.
-
Propongo externar a la paciente y continuar controles clínico e imagenológico de forma ambulatoria. Considero apropiado repetir RMI de cráneo y de médula en 3 meses.
BIBLIOGRAFÍA:
-
Dr. Javier Dutilh, “Manifestaciones neurológicas de enfermedades sistémicas (1° y 2° parte)” www.sitiomedico.org, Año 2002.
-
Francesc Graus, “A clinical approach to diagnosis of autoinmune encephalitis”; The lancet volume 15 N°4 p391-404, April 2016.
-
IntraMed, “Encefalitis autoinmune, abordaje clínico para su diagnóstico”, 04 Abril 2016.
-
Bartolomei S, “Medicina Interna, cálculos, scores y abordajes”. Segunda edición. Año 2010. Capítulo: Infectología, p724-726.
-
C. Sobrido Sampedro, “Neuroimagen del linfoma primario del sistema nervioso central en pacientes inmunodeprimidos.” Rev. Argentina de radiología. Vol. 78 N°1. Ciudad autónoma de Buenos Aires. Abril, 2014.
-
E. Graus Ribas; “Tumores intracraneales. Complicaciones neurológicas del cáncer.” Capitulo 176, p1461-1476. Farreras-Rozman. XVI edición. Volumen II.
-
A. J. da Rocha, “Linfoma primario del sistema nerviosos central: el aporte de las técnicas convencionales de diagnóstico por imágenes”. Rev Argent Radiol. Abril 2016; 80(2):112-121.
-
Jahnke K, Korfel A, O'Neill BP, et al.: “International study on low-grade primary central nervous system lymphoma”. Ann Neurol 59 (5): 755-62, 2006. (PUBMED Abstract).
-
Bromberg JE “CSF flow cytometry greatly improves diagnostic accuracyin CSN hematologic malignancies”. Neurology. 2007, May. 15;68(20):1674-9.
-
Alessandro, Lucas; “Estudio retrospectivo de 48 casos de linfoma primario del sistema nervioso central”. MEDICINA (Buenos Aires) Febrero 2017; 77: 17-23. Servicio de Neurología, Neurocirugia, Neuro-oncología, Medicina Interna. FLENI.
-
Farreras, Rozman; “Enfermedades desmielinizantes del sistema nerviosos central”. Cap. 177, p1467-1476. Vol. II, XVI Edición.
-
Hari. P, “Pulse corticosteroid therapy with methylprednisolone or dexamethasone” NCBI, PubMed1998, jul-aug; 65 (4): 557-60.
-
Dora Liu.; “A practical guide to the monitoringand managementof the complications of systemic corticosteroid therapy”. NCBI. Allergy Asthma Clin Immunol. Vol 9 (1):30, 2013 August.
-
Dr Leigh White, “Adult onset acute disseminated encephalomyelitis following appendicitis: a case report.” Journal of neurology and neuroscience ISSN 2171-6625. Vol.7 2016
-
Schwarzs S.; “Acute disseminated encephalomyelitis: a follow-up study of 40 adults patients”. NCBI. PubMed. Neurology. 2001, May 22; 56(10):1313-8
-
Neil Scolding; “Asociation of british Neurologist: revised (2015) guidelines for prescribing disease-modifying treatments in multiple sclerosis”. Journal pratical neurology 2015, 0: 1-7. Doi: 10. 1136
|
Imágenes del caso
|
