Discusión del caso clínico. | Presentación |
Como objetivos me propongo describir las cusas probables de debilidad muscular y oftalmoplejía en nuestra paciente, analizar diferentes enfermedades neuromusculares y repasar conceptos de miastenia gravis de presentación atípica.
DEBILIDAD MUSCULAR
Según su duración Los síntomas de debilidad muscular pueden ser intermitentes o persistentes. Los trastornos que ocasionan debilidad intermitente son:
-Miastenia gravis
-Parálisis periódicas: (hipopotasémica, hiperpotasémica, y paramiotonía congénita)
-Cuadros de déficit energéticos- metabólicos: de la glucólisis (déficit de miofosforilaza) y de la utilización de los ácidos grasos (déficit de carnitina palmitoil transferasa y de algunas miopatías mitocondriales). Las situaciones de déficit energético provocan rotura de las fibras musculares debidas a la actividad física, acompañadas de mioglobinuria.
La mayor parte de las enfermedades musculares causan debilidad persistente. En gran parte de ellas, incluidas casi todas las formas de distrofia muscular, polimiositis y dermatomiositis, los músculos proximales muestran mayor debilidad que los distales, y los músculos faciales se mantienen intactos, una tipología que se denomina de cintura. El diagnóstico diferencial se limita a otras características de la debilidad. La debilidad de los músculos de la cara y de la escapula alada son características de la distrofia facioescapulohumeral. La debilidad de los músculos de la cara y del área distal de las extremidades asociada con miotonía en la prensión manual es virtualmente diagnóstica de distrofia miotónica tipo I. Cuando hay debilidad en otros músculos inervados por pares craneales, con ptosis palpebral o debilidad de músculos extra oculares, los principales trastornos a considerar incluyen alteraciones de la unión neuromuscular (las enfermedades neuromusculares más importantes manifestadas con este patrón de debilidad incluyen: miastenia gravis, escleroris lateral amiotrofia, miopatía nemalínica de inicio tardío, hiperparatiroidismo, miositis focal, y algunas formas de miopatías con cuerpos de inclusión), distrofia oculofaríngea, miopatías mitocondriales o alguna miopatía congénita.
Todo trastorno que cursa con debilidad muscular se puede acompañar de fatiga, dada la imposibilidad de mantener la fuerza.
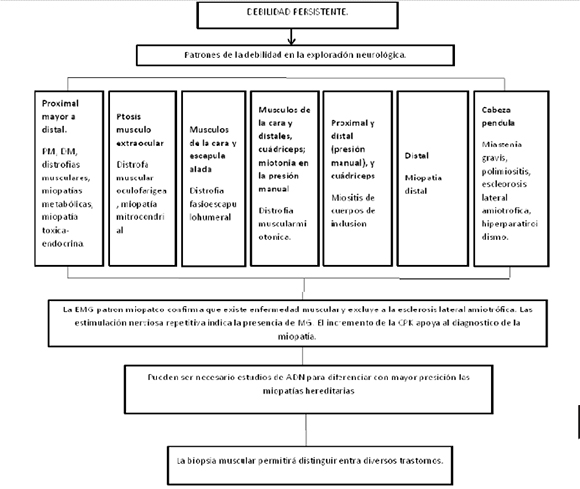
De acuerdo con la topografía de la lesión, se las puede diferenciar en enfermedades de afectación central como la enfermedad de Wernike, infarto pontino, glioma infiltrante, esclerosis múltiple, encefalomielitis aguda diseminada; las cuales se encuentran claramente descartadas en nuestra paciente por no ser la clínica ni estudios por imágenes compatibles; afectación de nervios centrales como ser: síndrome de Guillain Barré, meningitis neoplásicas o granulomatosas, trombosis de senos cavernosos, síndrome de Tolosa Hunt, pseudotumor orbitario, también descartadas en nuestro paciente por la clínica incompatible. Otras patologías se presentan con afectación de placa neuro-muscular como la miastenia gravis, síndromes miasténicos congénitos, síndrome de Lambert Eaton, oftalmopatía tiroidea, botulismo, neuropatías hereditarias sensitivo-motoras. Por último, otras enfermedades se producen por afectación muscular: oftalmoplejía externa progresiva, distrofias musculares, polimiopatías congénitas, miopatías, parálisis periódicas familiares, enfermedades musculares inflamatorias. Por las características clínicas de nuestra paciente, se plantean como causas principales trastornos de la placa neuro-muscular o muscular puro.
Entre las enfermedades MUSCULARES, las miopatías inflamatorias constituyen el principal grupo de causas adquiridas y potencialmente curables de debilidad muscular. Se clasifican en estos tres grupos principales: POLIMIOSITIS, DERMATOMIOSITIS Y MIOSITIS POR CUERPOS DE INCLUSION. Evolucionan como enfermedades progresivas simétricas excepto la miositis de cuerpos de inclusión, que puede tener una presentación asimétrica. Los movimientos motores finos que dependen de la fuerza de los músculos distales, son solo afectados en las fases tardías. Los músculos oculares están respetados, incluso en los casos avanzados. La musculatura facial tampoco se ve afectada. En los casos no tratados, la debilidad grave casi siempre se vincula con la atrofia muscular. La sensibilidad esta conservada, y los reflejos osteotendinosos también están preservados, aunque en los músculos afectados con atrofia importante pueden desaparecer.
Las distrofias, las cuales considero podrían estar relacionadas con el cuadro de nuestra paciente son:
-
La distrofia muscular oculofaríngea, enfermedad progresiva que aparece en la edad adulta (entre 40 y 60 años) y se caracteriza por ptosis y disfagia. Su evolución es lenta con agravación progresiva de las limitaciones funcionales.
-
Las distrofias musculares de cinturas (LGMD), el grupo más heterogéneo desde el punto de vista clínico como molecular. Inicialmente se agruparon bajo este nombre aquellas distrofias musculares con debilidad fundamental de la cintura pelviana o escapular, y que no correspondían al fenotipo Duchenne o facioescapulohumeral. Se dividen en dos grandes grupos según su modo de herencia autosómico dominante (LGMD1) o recesivo (LGMD2). Las formas graves suelen corresponder al grupo de las recesivas, como las debidas al déficit de sarcoglicanos, calpaína o disferlina. En el grupo de las dominantes existen formas más leves, incluyendo hiperCKemia asintomática (como sucede en algunas formas por déficit de caveolina-3).
-
La distrofia muscular facioescápulohumeral, una de las distrofias musculares más frecuentes, y tiene un patrón de herencia autosómico dominante. Suele manifestarse en la juventud, aunque existen formas de inicio más tardío y la penetrancia es variable. Se caracteriza por la presencia de debilidad y atrofia de los músculos de la cara y de la cintura escapular: movilidad facial reducida, dificultad para levantar los brazos por encima de la cabeza, hombros caídos hacia delante y omóplatos prominentes. Los glúteos y músculos anteriores de la pierna están afectados. Su evolución es muy lenta con frecuentes períodos de estabilización. La esperanza de vida es normal a pesar de que la incapacidad funcional es, a menudo, grave. A esta última la considero un diagnostico deferencial a tener en cuenta.
Un diagnóstico a considerar en este caso es la oftalmoplejía externa progresiva crónica (CPEO), una enfermedad mitocondrial común. Este grupo de enfermedades presenta solapamiento clínico, enzimático y genético entre las diferentes entidades. La ptosis suele ser la primera evidencia clínica y puede preceder a la oftalmoparesia en meses o años. Dicha oftalmoparesia se caracteriza porque progresa de forma lenta y suele ser bilateral. Ante la sospecha diagnóstica de oftalmoplejía externa progresiva crónica se solicita estudio con pruebas complementarias, obteniendo la confirmación etiológica del cuadro clínico con la biopsia muscular que pone de manifiesto la presencia de fibras rojo rasgadas musculares. La mayoría de los casos de OEPC son esporádicos, causados por deleciones en el ADN mitocondrial. Es importante realizar el diagnóstico diferencial con otros cuadros clínicos que asocian ptosis y oftalmoparesia como la miastenia gravis. La biopsia muscular es la herramienta más útil para su diagnóstico. La mayoría de los pacientes no requieren cirugía de estrabismo y la cirugía palpebral se indica para los casos con ptosis severa.
Otras de las entidades que pueden estar relacionadas con nuestro posible diagnóstico, son aquellas que afectan a la UNION NEURO-MUSCULAR.
Afectan a la musculatura y al sistema nervioso, pudiendo estar comprometidos: la unión neuromuscular, el nervio periférico (en brazos, piernas, cuello y cara), o la motoneurona espinal. Su aparición puede producirse en cualquier etapa de la vida, tanto en el nacimiento como en la adolescencia o en la edad adulta.
Se encuentran dentro del grupo de las denominadas enfermedades raras, son enfermedades poco conocidas y son muy diversas.
MIASTENIA GRAVIS DE PRESENTACIÓN ATÍPICA:
La Miastenia Gravis es una enfermedad de causa inmunológica, producida por la presencia de anticuerpos contra componentes de la membrana post sináptica de la unión neuromuscular; en la mayor parte de los casos el paciente desarrolla anticuerpos contra los receptores de acetilcolina (AchR) o los receptores de la tirosinquinasa músculo específicos (Anti-musk). Es una enfermedad que puede manifestarse a cualquier edad, aunque lo más frecuente es entre los 20 y 30 años en las mujeres y entre los 40 y 60 en los hombres. Se caracteriza por presentar una debilidad muscular de intensidad y duración variables que pueden afectar a cualquier músculo. Esta debilidad puede aumentar con el esfuerzo y/o con la repetición del movimiento. Su evolución es variable, con remisiones o exacerbaciones. Se trata con inhibidores de la acetilcolinesterasa e inmunosupresores. En ciertos casos en necesaria la timectomía.
Hago especial mención a los diferentes tipos de miastenia gravis, los cuales considero más cercanos al posible diagnóstico de la paciente:
Miastenia de afectación ocular: los síntomas y signos están limitados a los músculos oculares (elevador del párpado y extraoculares). Se evidencia ptosis y diplopía en el 85% de los casos y si la debilidad permanece limitada por más de dos años a estos músculos, cabe un 90% de posibilidad de que la miastenia no se generalice. El diagnostico se lleva a cabo con electromiografía, especialmente la de fibra única.
Miastenia seronegativa: entidad en la cual los anticuerpos suelen no manifestarse, y si lo hacen, en general los anti receptor de acetilcolina son negativos y solo en un 40% de los casos son positivos los anti receptores de tirosinquinasa musculo específicos. Suele tener una presentación con clínica respiratoria, axial y bulbar. Es refractaria al tratamiento convencional en la mayoría de los casos.
Aunque más alejado de nuestro posible diagnóstico, otra enfermedad a tener en cuenta es el síndrome de Lambert-Eaton, una enfermedad debida a anticuerpos contra componente de la membrana pre sináptica de la unión neuromuscular que afecta principalmente a los canales de calcio. En un porcentaje elevado de las ocasiones se trata de una enfermedad paraneoplásica, por lo que se debe descartar la presencia de un cáncer oculto (especialmente de pulmón). Produce debilidad en MMII, ptosis, hipo y arreflexia y alteraciones autonómicas (impotencia, estreñimiento). Se llega al diagnostico mediante electromiografía, y el tratamiento suele ser Piridostigmina, plasmaféresis o inmunosupresores.
Síndromes miasténicos congénitos: se trata de enfermedades genéticamente determinadas, que aparecen desde el nacimiento. Se caracterizan por la presencia de fatiga anormal debida a una debilidad muscular localizada o generalizada. Algunas formas responden de forma parcial a tratamiento con anticolinestrásicos. Existe una forma adulta de posible comienzo tardío (el síndrome del canal lento).
Para culminar, creo oportuno, aunque claramente alejado una vez obtenidos los anticuerpos correspondientes, nombrar brevemente a la OFTALMOPLEJÍA TIROIDA o Enfermedad de BASEDOW como otra entidad a descartar. Causada por una activación inadecuada del sistema inmunológico (autoanticuerpos) que elige como blanco a los receptores de TSH de las células foliculares, resultando la síntesis y secreción excesiva de hormona tiroidea.
De acuerdo con el caso, y tras la obtención de resultados tales como: espirometría, tomografía de tórax y resonancia magnética de cráneo y anticuerpos anti tiroideos normales, y con un resultado parcial al tratamiento con piridostigmina, propongo considerar como primeras hipótesis diagnósticas a la miastenia gravis de presentación atípica (seronegativa y de afectación ocular pura) sin dejar de considerar también a la oftalmoplejía externa progresiva como otro probable diagnóstico con fuerte peso en nuestra paciente. Mucho más alejado pero posible también, a la distrofia óculo-faríngea; para lo cual me parece oportuno contar con los resultados finales de las serologías virales, el informe final de la electromiografía, la cual orientara a un diagnóstico específico y esperar los resultados de los anticuerpos Anti muSk y AchR. Como interrogante final me planteo si continuar o no con el tratamiento instaurado, creyendo oportuno reevaluación a la brevedad.
Bibliografía:
-
Jimenez- Caballero PE, Serviá M. Oftalmoplejía externa progresiva crónica: manidestaciones clinicas y electromiograficas en una serie de casos. Revista de neurologia 2006;43:724-728.
-
Harrison principios de medicina interna, 18° edición, Miastenia grave y otras enfermedades de la unión neuromuscular, sección 3, capítulo 386.
-
Harrison principios de medicina interna, 18° edición, Distrofias musculares, y otras enfermedades musculares, sección 3, capítulo 387.
-
Gregorio Arellano-Aguilar, Erik Santiago Núñez-Mojica. Paraneoplastic Lambert-Eaton syndrome in a patient with disseminated metastatic cáncer. Cirugía y cirujanos. 2018;86:79-83
|
Imágenes del caso
|
