Discusión del caso clínico. | Presentación |
Objetivos:
-
Establecer principios básicos sobre el abordaje de pancitopenias.
-
Considerar causas de esplenomegalia.
-
Considerar causas de infarto esplénico.
-
Considerar posibles diagnósticos en este paciente.
-
Consideraciones finales del caso.
Pancitopenias
Al hablar de pancitopenias nos referimos a la presencia simultánea de anemia (hemoglobina menor a 13g% en hombres y menor a 12 g% en mujeres), leucopenia (recuento de leucocitos menor a 4500/mm3) y trombocitopenia (recuento de plaquetas menor a 150.000/mm3).
Las pancitopenias pueden clasificarse de la siguiente manera:
-
Centrales o periféricas: según exista disminución de las células hematopoyéticas en la medula ósea o un descenso periférico de los elementos formes de la sangre con médula ósea normal o no y en este caso será por destrucción (pancitopenias autoinmunes) o por secuestro (hiperesplenismo).
-
Hereditarias o adquiridas: la patogenia en los fallos medulares primarios consiste en la disminución de la hemopoyesis debido a una enfermedad primaria de la médula ósea, en cuya etiopatogenia intervienen alteraciones genéticas de las células madre hemopoyéticas y fenómenos de autoinmunidad. Siempre habrá que descartar previamente carencias de nutrientes, toxicidad por drogas, químicos o radiaciones, enfermedades neoplásicas, metabólicas o inflamatorias, que pueden afectar la hematopoyesis.

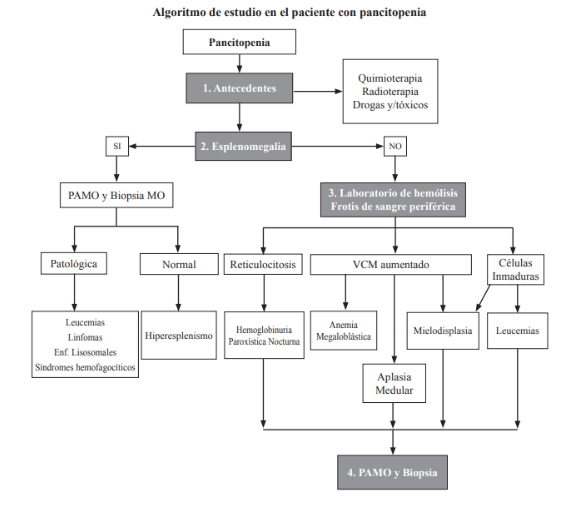
El abordaje de las pancitopenias requiere de:
-
Historia clinica. Presencia o no de síntomas. Hepatoesplenomegalia. Adenopatias.
-
Metodología diagnóstica.
-
Hemograma completo con fórmula y plaquetas. Velocidad de eritrosedimentación. Función hepática. LDH. Reticulocitos.
-
Frotis de sangre periférica.
-
Punción aspiración de médula ósea. Biopsia de médula ósea.
Esplenomegalia
El bazo es el órgano linfático más grande del organismo. Cuyas funciones son hematopoyética, hemocaterítica, inmunológica, de aclaramiento (sistema retículo endotelial) y de reserva de elementos sanguíneos (p.ej. almacena un 20 a 30% de todas las plaquetas).
Se define esplenomegalia como el incremento del tamaño de bazo mayor a sus dimensiones normales, (en el adulto son 12 × 7 × 3.5 cm) con un peso aproximado de 150 gramos y un volumen de 300 ml. Los mecanismos por los cuales se produce esplenomegalia consistente en aumento de los folículos linfoides, hiperplasia del sistema retículo endotelial, infiltración por células neoplásicas, estasis vascular (p.ej. hipertensión portal) o exacerbación de función fisiológica del bazo como ocurre en hematopoyesis extramedular.
Las principales causas de esplenomegalia pueden abordarse en 4 grupos:
- Infecciosas: fiebre tifoidea, brucelosis, endocarditis infecciosa, mononucleosis, paludismo y kala azar.
- Hepáticas: hepatopatía aguda y crónica, trombosis de vena esplénica, portal o síndrome de Budd Chiari.
- Hematológicas: síndromes linfoproliferativos, síndromes mieloproliferativos, anemias hemolíticas congénitas como esferocitosis o talasemia o enfermedades del sistema mononuclear fagocítico como enfermedades de depósito o histiocitosis.
- Misceláneas: amiloidosis, sarcoidosis y colagenosis como lupus eritematoso o artritis reumatoidea.
Para el estudio de la esplenomegalia serán fundamentales:
- Historia clínica. Antecedentes personales y familiares. Examen físico.
- Pruebas por imágenes: ecografía, tomografía computarizada o resonancia magnética.
- Laboratorio: hemograma completo con fórmula y plaquetas e índices hematimétricos, reticulocitos, pruebas de función hepática y LDH. Frotis de sangre periférica. Laboratorio inmunológico.
- Otros: Cultivos. Serologías virales (virus hepatitis B y C, virus de inmunodeficiencia humana, VEB y CMV). Estudio de trombofilias. Mutación JAK2. Aspirado y biopsia de médula ósea.
Se define esplenomegalia gigante o masiva como al crecimiento del bazo cuyo polo inferior de encuentra hasta la pelvis, o cuando cruza la línea media hacia los cuadrantes abdominales derecho e inferiores. Dentro de sus principales causas deben citarse al paludismo, kala azar, quiste hidatídico, talasemia mayor, enfermedad de Gaucher, mielofibrosis primaria, leucemia prolinfocítica, tricoleucemia y leucemia mieloide crónica.
Las principales complicaciones son:
- Hiperesplenismo. El mismo se caracteriza por la presencia de esplenomegalia, citopenias periféricas, médula ósea normal o hiperplásica y corrección de la citopenia tras esplenectomía.
- Ruptura esplénica (espontánea o tras traumatismos menores).
- Infarto esplénico. Los mismos ocurren cuando la arteria esplénica o alguna de sus ramas se ocluyen por un émbolo séptico o por un trombo. Clásicamente cursan con dolor agudo en hipocondrio izquierdo. Pueden ocurrir en diferentes situaciones: estados de hipercoagulabilidad (tumores, síndrome antifosfolípido), patologías embolígenas (fibrilación auricular, foramen oval permeable, ateromatosis, endocarditis infecciosa), embolización terapéutica de la arteria esplénica, tumores hematológicos asociados con esplenomegalia como mielofibrosis o leucemia mieloide crónica, hemoglobinopatías (especialmente la anemia de células falciformes), o cualquier patología que curse con esplenomegalia (enfermedad de Gaucher, linfoma primario esplénico), traumatismos esplénicos que comprometan su vascularización, torsión de la arteria esplénica, incluso como complicación en una mononucleosis. El tratamiento depende de la causa. Los casos más sencillos requieren medicación analgésica y de soporte, mientras que complicaciones como el absceso o la ruptura pueden requerir cirugía.
En el caso de nuestro paciente deberá considerarse las siguientes etiologías:
1. Linfoma primario de bazo.
Es una entidad inusual del bazo que comprende menos del 2% de todos los linfomas y el 1% de los linfomas no Hodgkin. Implica el compromiso limitado a bazo o ganglios linfáticos hiliares sin recurrencia tras 6 meses de esplenectomía y sin presencia de otro linfoma, especialmente en el hígado o en los nódulos linfáticos para-aórticos o mesentéricos. La edad media de presentación es de 36 años, con un rango entre los 22 y los 48 años. Es más frecuente en la mujer con una relación de 4:1.
Hay dos tipos principales:
- Linfoma esplénico con linfocitos vellosos circulantes.
- Linfoma esplénico de la zona marginal.
Las manifestaciones clínicas más descriptas son:
-
Síntomas inespecíficos como pérdida de peso, debilidad, fiebre, dolor abdominal o malestar en contexto de esplenomegalia.
-
Síntomas específicos como resultado de la invasión de órganos adyacentes como estómago, páncreas, diafragma, colon o mesenterio.
-
Excepcionalmente son asintomáticos.
Dichos hallazgos se encuentran asociados a pancitopenia con aumento de reactantes de fase aguda y LDH. A través de la evaluación por frotis de sangre periférica se constatan células linfoides neoplásicas como células peludas, prolinfocitos, linfocitos vellosos, linfocitos vellosos basófilos, etc. El diagnóstico debe realizarse en tejido ganglionar o extraganglionar obtenido preferentemente por biopsia escisional. Las biopsias por tru-cut pueden ser suficientes cuando no se tenga tejido accesible. La biopsia por aspiración con aguja fina (BAAF) puede sugerir el diagnóstico. Deberá realizarse Inmunohistoquímica mínima obligatoria: CD45, CD20 y CD3. El reporte histopatológico debe realizarse de acuerdo a la clasificación vigente de la OMS.
En la actualidad la ecografía (US) y la tomografía axial computarizada (TAC) son las modalidades más usadas para el diagnóstico, pues con estas técnicas es relativamente fácil su detección. En un principio debe realizarse el diagnóstico diferencial con infecciones bacterianas, colagenosis y otros procesos tumorales de localización esplénica, pero el diagnóstico definitivo requiere de la esplenectomía y posterior estudio anatomopatológico. La resonancia magnética está indicada fundamentalmente en sistema nervioso central, evaluación ósea y fundamentalmente de la médula espinal.
La tomografía por emisión de positrones (PET) es capaz de demostrar enfermedad activa en ganglios aumentados de tamaño y también en ganglios de tamaño normal. Se han descripto cuatro patrones radiológicos en la tomografía axial computarizada (TAC) helicoidal en el LPB. No obstante, este patrón radiológico no es específico del linfoma primario esplénico, puede encontrarse también en metástasis, abscesos, hematomas, angiosarcomas y hamartomas.
La infección por el virus de la hepatitis C se ha asociado a diferentes tipos de linfomas no Hodgkin, incluyendo el linfoma de zona marginal (esplénico, nodal y extranodal). En la bibliografía médica, se ha publicado además, un linfoma esplénico primario (LPB) relacionado con el virus de la hepatitis B (VHB).
Como medidas terapéuticas se deberá realizar esplenectomía que debe ser seguida de terapia anticoagulante y quimioterapia.
2. Linfoma hepatoesplénico de células T (HSTL).
Este linfoma no Hodgkin constituye una rara variante de linfomas de células T periféricos extranodales con infiltración de hígado, bazo y médula ósea. Representa tan solo el 3% de los subtipos de linfoma de células T en los Estados unidos. Presenta un curso de enfermedad agresiva y un registro implacable de resultados letales. Suele presentarse en hombres jóvenes o en pacientes con inmunodepresión crónica durante la tercera a cuarta década de vida. De las causas conocidas, la desregulación inmune y la inmunosupresión son clave en la patogénesis. Las manifestaciónes clínicas incluyen hepatoesplenomegalia y síntomas constitucionales. La afectación de la médula ósea u organomegalia pueden causar citopenias.
El diagnóstico generalmente se realiza después de la esplenectomía o biopsia de hígado/médula ósea con inmunofenotipificación adecuada. La mayoría de los casos han mostrado tinción positiva para el TCR γδ o, con menor frecuencia, TCR αβ. Los regímenes basados en antraciclinas proporcionan respuestas modestas con la mayoría de los individuos que mueren dentro de un año del diagnóstico. El trasplante de células madre hematopoyéticas es una opción terapéutica.
3. Síndromes linfoproliferativos crónicos.
Se caracteriza por la proliferación clonal de linfocitos de morfología madura pero función anormal. La mayoría (<85%) de los síndromes linfoproliferativos crónicos son de línea B encontrándose dentro de esta entidad a la leucemia linfática crónica, leucemia prolinfocítica B y tricoleucemia o leucemia de células peludas. Debido a que la edad de presentación (>50 años) y los hallazgos analíticos (leucocitosis) no coinciden con nuestro paciente nos centraremos exclusivamente en la tricoleucemia.
La tricoleucemia es una neoplasia linfoide infrecuente caracterizada por la acumulación de linfocitos B anormales. La anomalía más frecuente es la tricitopenia asociada a la monocitopenia. En un 80 a 90% se produce esplenomegalia que puede ser masiva, mientras que las linfoadenopatias periféricas son poco frecuentes (<5%). La edad media de diagnóstico son los 60 años pero no son infrecuentes los casos en menores de 40 años. Ocasionalmente existe enfermedad extramedular/esplénica. Pueden ocurrir complicaciones esqueléticas (3%) como lesiones líticas con afectación del fémur proximal o afecciones autoinmunes.
La morfología característica en sangre o médula ósea es la de células linfoides grandes (duplican el tamaño de un linfocito de leucemia linfática crónica) con amplio citoplasma con proyecciones similares a pelos en aproximadamente el 85% de los casos. Históricamente el diagnóstico se confirmó mediante la tinción TRAP (fosfatasa ácida tartrato resistente). Como la aspiración de médula ósea es efectiva solo en el 10% (aspirado seco), la biopsia de médula ósea es indispensable. El inmunofenotipo es fundamental ya que a través de citometría de flujo se determina la positividad para CD11c, CD19, CD20, CD22, CD25, CD123 y FMC7 y negatividad para CD5 y CD23.
Histológicamente se caracteriza por la infiltración de la pulpa roja diferenciándolo de otros síndromes linfoproliferativos de células B.
Se iniciará tratamiento ante citopenias sintomáticas o esplenomegalia dolorosa siendo la primera recomendación terapéutica la cladribina.
4. Síndromes mieloproliferativos.
Se caracterizan por la alteración clonal en una célula stem con afectación de la serie mieloide presentando panmielosis en sangre periférica asociado a hematopoyesis extramedular (hepatoesplenomegalia). En relación a esplenomegalia masiva debería considerarse la leucemia mieloide crónica y la mielofibrosis primaria sin embargo la edad de presentación (entre 50 a 60 años) asociado a la ausencia de alteraciones analíticas (ej. leucocitosis) o en frotis de sangre periférica (ej. dacriocitos o reacción leucoeritroblástica en la mielofibrosis primaria) hacen improbables estos diagnósticos.
5. Enfermedades de depósito (tesaurismosis).
Existe un error en el metabolismo celular, de modo que falta o hay un trastorno funcional en una enzima generando la acumulación (generalmente en lisosomas) del metabolito que en condiciones normales es metabolizado por esa enzima. En este grupo se incluye a la enfermedad de Gaucher, enfermedad de Niemann- Pick y enfermedad de Fabry.
En relación a nuestro paciente debería considerarse la enfermedad de Gaucher que se trasmite por herencia autonómica recesiva producto del déficit de B glucocerebrosidasa generando la acumulación de glucocerebrosido en médula ósea, bazo, hígado, pulmón, tejido esquelético y en las formas neurológicas en cerebro. Presenta una prevalencia de 1/40.000 a 1/60.000 personas salvo en los judíos Ashkenazi en quienes es cien veces mayor. Se clasifica en tres formas clínicas según la ausencia (Tipo I) o presencia (tipo 2 y 3) de signos neurológicos. La presentación puede ser en la infancia (Tipo 2 y 3), jóvenes o adultos (tipo 1). Clínicamente se caracteriza por visceromegalias, citopenias y afectación ósea debido a la infiltración en medula ósea de las células de Gaucher que estimulan a la fibrosis, necrosis, infartos, lesiones líticas, osteopenia, deformidades y fracturas patológicas. Aunque 10 a 25 % de los pacientes son asintomáticos. Es importante realizar el diagnostico precoz porque los pacientes pueden beneficiarse de la terapia de reemplazo enzimático.
6. Mieloptisis.
La mieloptisis se define como la infiltración de la médula ósea por células no hematopoyéticas, lo que produce desplazamiento de la hematopoyesis normal, generando diferentes grados de anemia, trombocitopenia y neutropenia.
La presencia de células tumorales metastásicas en el espacio medular, es un fenómeno que ocurre hasta en el 30% de los pacientes con neoplasias avanzadas.
Las distintas series de casos evidencian que los tumores que se relacionan con esta entidad son en mayor medida: cáncer de pulmón, mama, próstata, tiroides, riñón, primario desconocido. En menor medida: cáncer gástrico, tumores germinales y cáncer de cérvix. Se podrá visualizar a través del frotis de sangre periférica la presencia de reacción leucoeritroblástica caracterizada por la presencia de eritrocitos nucleados y con una desviación a la izquierda en los neutrófilos en sangre periférica.
La demostración de mieloptisis en pacientes oncológicos se asocia con un pronóstico ominoso, especialmente si se tiene en cuenta que esta complicación se hace manifiesta cuando las células tumorales desplazan más del 60% de las células hematopoyéticas normales. En término generales, después de la certificación de esta entidad, el 50% de los pacientes tienen una supervivencia global inferior a 2,5 meses. El pronóstico es especialmente malo en quienes debutan con un nivel de hemoglobina inferior a 8,5 mg/dl, en aquellos que presentan tres o más sitios de metástasis (especialmente si son viscerales), en quienes tienen neutropenia febril, en aquellos que tienen fosfatasa alcalina mayor de 800 mg/dl y/o LDH mayor a 1000 UI/ml.
En el caso de nuestro paciente los estudios por imágenes solicitados (tomografía computada de tórax y abdomen y ecografía testicular) junto a la citometría de flujo descartan este diagnóstico.
7. Leucemias agudas.
Las leucemias agudas se caracterizan por la proliferación incontrolada de células inmaduras (blastos) que infiltran la médula ósea > 20% generado un desplazamiento de la hematopoyesis normal.
Las mismas pueden clasificarse como:
Primarias o secundarias debido a la evolución de otras hemoglobinopatías ej. Anemia de Fanconi y síndrome mielodisplásico o carcinógenos.
Leucemias mieloblásticas agudas (clon proliferante de origen mieloide) o linfoblástica aguda (clon proliferante de origen linfoide).
Las leucemias pueden ser secundarias por ejemplo a síndromes de fallo medular hereditario que son enfermedades genéticas raras caracterizadas por diversos grados de déficit en la producción de eritrocitos, granulocitos y plaquetas en la médula ósea. Dentro de este haremos hincapié en la anemia de Fanconi que es un desorden genético de herencia autosómico recesiva y en raros casos ligado al cromosoma X y fenotípicamente heterogéneo caracterizado por anomalías congénitas, fallo medular progresivo y progresión al desarrollo de leucemias y otras formas de cáncer. La prevalencia estimada es de 10 casos por millón de individuos con una mediana de edad al diagnostico de 7 años. Siendo más frecuente en varones. La ocurrencia de malformaciones físicas, la edad de aparición de aplasia, leucemia o cáncer dependen del genotipo, de la penetrancia de cada mutación y de su expresión.
Se caracteriza por:
Fallo medular progresivo.
Las anomalías congénitas son frecuentes destacándose anomalías cráneo faciales (microcefalia, cara triangular o dismorfias), esqueléticas (pulgar, radio, cubito, mano anormales o luxación congénita de cadera, anomalías de pies y piernas, deformidades óseas, espina bífida o malformaciones vertebrales), cardiovasculares (ductus persistente), renales (riñón ectópico en herradura o hipoplásico), neurológicas (pituitaria pequeña o ausencia de cuerpo calloso), piel (manchas café con leche o hiperpigmentación), talla baja, genitales (hipogonadismo, criptorquidia, anomalías genitales internas y externas), ojos (microftalmia), oídos (sordera) o gastrointestinal (atresia, meckel o ano imperforado). Sin embargo un 25% presentara fenotipo normal.
Endocrinopatías (81%) ej. diabetes mellitus, insuficiencia de la hormona de crecimiento, hipotiroidismo, hipogonadismo y osteopenia/osteoporosis.
Predisposición a enfermedad maligna. El riesgo de padecer cáncer es 800 a 1000 veces mayor que la población general siendo más común LMA, carcinomas de células escamosas, tumores de cerebro y tejidos blandos. Las leucemias en estos pacientes suelen ser LMA (de MO a M7 excepto M3) aunque se han descripto casos de LLA y LMMC.
Su diagnóstico requiere de test genéticos para mutaciones de genes FANC. Una vez diagnosticado se realizará tratamiento médico escalonado basado en andrógenos, citoquinas y régimen transfusional.
Considero que nuestro paciente presenta alteraciones como hepato esplenomegalia o lesiones osteolíticas que no coinciden con la posibilidad de presentar anemia de Fanconi.
Posteriormente se realizo citometría de flujo en la que se constato una Leucemia linfoblástica aguda tipo B común en un 7.5%.
La leucemia linfoblástica aguda resulta de la proliferación clonal de linfoblastos en médula ósea o sangre periférica >20% (OMS 2016). Presenta una distribución bimodal con un primer pico en pacientes menores de 20 años y el segundo a partir de los 45 años de edad. Representa el 75 a 80 % de las leucemias agudas en edad pediátrica, predominando entre los 2 a 5 años. La misma representa menos del 1% de las neoplasias en los adultos, siendo aproximadamente cuatro veces menos frecuentes que en niños.
Las mismas pueden ser T (15 a 20%) o B siendo estas ultima la más frecuente como es en el caso de nuestro paciente.
Clínicamente se manifiesta de 3 formas:
Síntomas constitucionales: cansancio, debilidad, pérdida de peso, sudoración nocturna, etc.
Síntomas por infiltración de la médula ósea: anemia, leucopenia y hemorragias.
Síntomas por infiltración de otros órganos:
Dolor óseo espontaneo o a la presión (infiltración medular) y articular en el 21 a 59% de los niños, mientras que sólo el 4% de los adultos presentan manifestaciones musculoesqueléticas.
Adenopatías (generalmente cervicales) y esplenomegalia moderada (70 a 80%) o hepatomegalia.
Infiltración de piel, mediastino, sistema nervioso central o testículo.
Los hallazgos analíticos consisten en blastos de estirpe linfoide en sangre periférica (leucocitosis) e infiltración de la médula ósea con presencia de pancitopenia y aumento del turn-over celular (LDH y ácido úrico). Siempre deberá realizarse evaluación del sistema nervioso central y testicular.
Dentro de los factores de riesgo adversos en adultos se encuentran la edad (a mayor edad peor pronóstico), recuento leucocitario (>30.000/mm3 en línea B), inmunofenotipo LLA pro B, determinadas alteraciones citogenéticas y moleculares (ej, cromosoma Philadelphia +), falta de respuesta precoz a la terapia de inducción, etc.
El tratamiento de adultos menores a 60/65 años con cromosoma Philadelphia negativo se divide en fases que incluyen inducción (4 a 6 drogas: vincristina, antraciclinas, corticoides, asparaginasa, ciclofosfamida, citarabina y 6 mercaptopurina); consolidación (metotrexato a altas dosis o fraccionado, citarabina, asparaginasa y 6 mercaptopurina) y mantenimiento prolongado (6 mercaptopurina continuo y metotrexato semanal). Todos los esquemas de tratamiento incluyen profilaxis o tratamiento del sistema nervioso central. Mientras que aquellos con cromosoma Philadelphia positivo requerirán de inhibidores de la tirosina quinasa y quimioterapia.
En relación a nuestro paciente se han descripto reportes bibliográficos sobre presentación de LLA-B con lesiones osteolíticas múltiples con hipercalcemia y ausencia de blastos en sangre periférica siendo muy raros en adultos. Las anomalías esqueléticas incluyen osteoporosis, reacción perióstica, esclerosis reactiva, defectos líticos y fracturas vertebrales por compresión. Cabe destacar que esta forma de presentación no implica por si sola un pronóstico adverso.
Consideraciones finales:
Nos encontramos frente a un paciente masculino de 21 años quien presentaba pancitopenia con neutropenia febril, hepatoesplenomegalia, infarto esplénico y lesiones osteolíticas donde para el esclarecimiento del cuadro fue fundamental la realización de punción aspiración de médula ósea y a través de la citometría de flujo se llego al diagnostico de leucemia linfoblástica aguda tipo B común en un 7.5% por lo que considero fundamental los resultados anatomopatológicos de la biopsia de médula ósea y pesquisar alteraciones citogenéticas/moleculares para definir pronóstico e iniciar tratamiento quimiotérapico oportuno.
Bibliografía
Jesús F. San Miguel. Fermín M. Sánchez Guijo. Hematología manual básica razonada 3° edición. 2009
Pablo Vargas Viverosa, Rafael Hurtado Monroya , José Ángel Villalobos Alvab. Esplenomegalia. Monografía Scielo. Vol. 56, N.o 2. Marzo-Abril 2013
ROJO ALVARO, J. et al. Lesiones esplénicas en medicina interna. Anales Sis San Navarra, Pamplona , v. 37, n. 1, p. 169-176, abr. 2014.
MAS MEDINA, Vicente Joaquín et al. Linfoma primario de bazo. Presentación de un caso. Gac Méd Espirit, Sancti Spíritus , v. 15, n. 3, p. 317-323, dic. 2013.
Sachin B. Chitra .R. Primary splenic lymphoma: Current diagnostic trends. World journal of clinical cases. World J Clin Cases. 2016 Dec 16; 4(12): 385–389.
Elena, Graciela Milovic, Vera Ramos, Anahí Rossi, Blanca de los Milagros Touliet, Valeria. Sindrome de falla medular. Sociedad argentina de hematología. Guías de Diagnóstico y Tratamiento 2017.
Bezares, Raimundo Bistmans, Alicia Borge, Mercedes Cabrejo, Maria del Rosario Custidiano, Rosario Dupont, Juan Ferini, Gonzalo Fernandez Grecco, Horacio Gamberale, Romina Giordano, Mirta Kornblihtt, Laura Kruss, Mariana Miroli, Augusto Pavlovsky, Miguel Risveros, Dardo Rodríguez, Cecilia Slavutsky, Irma. Síndromes linfoproliferativos crónicos. Sociedad argentina de hematología. Guías de diagnostico y tratamiento 2017.
Bendek, Georgina Elhelou, Ludmila Ferrari, Luciana Heller, Paula Kornblihtt, Laura Larripa, Irene Mela Osorio, María José Moiraghi, Elena Beatriz Molinas, Felisa Narbaitz, Marina Pérez, Mariela Schepps, Karen Rojas, Francisca Roveri, Eriberto Sackmann, Federico Sánchez Ávalos, Julio Varela, Ana Inés Vijnovich Baron, Anahí. Neoplasias mieloproliferativas crónicas clásicas BCR-ABL negativas. Sociedad argentina de hematología. Guías de diagnostico y tratamiento 2017.
Javier Severini, Carolina Tardío, Mariana Bellantig Tardío, Mariana Cusumano, Vanesa Dolce, Daniela Perotti, Georgina Grossi, Fabián Trivisonno, Julio Miljevic. Hospital Juan Bautista Alberdi. Rosario. Abordaje del paciente con pancitopenia.2010
Maria Colquicocha-Murillo, Janetliz Cucho-Jurado, Renee Mercedes Eyzaguirre-Zapata, Gioconda Manassero-Morales, Mariela del Carmen Moreno-Larrea, Katia Liliana SalasArbizu, Aimee Margarita Torres-Argandoña y Jesús Olga Vargas-Castro. Guía para diagnóstico y tratamiento de la Enfermedad de Gaucher. Rev Med Hered. 2015; 26:103-121.
Mauricio Luján y Medellin Andrés. Mieloptisis. Acta médica colombiana. Vol.34 N°4. Octubre diciembre 2009.
Agriello, Evangelina Cazap, Nicolás Dourisboure, Ricardo (✝) Fernández, Isolda Ferrari, Luciana Fischman, Laura Funes, María Eugenia Giménez Conca, Alberto González, Jacqueline Lang, Cecilia Mela Osorio, María José Moirano, María Mercedes Oliveira, Natalia Rey, Irene Riccheri, Cecilia Zanella, Lorena. Leucemias agudas. Sociedad argentina de hematología. Guías de diagnostico y tratamiento 2017.
González S, Beccacece M, Sardu L, Avila L. leucemia linfoblástica aguda: rara presentación con lesiones osteolíticas múltiples. Sociedad argentina de hematología. Hematología. Volumen 21 N°3: 310-316,2017.
Sima Amin , Sarah K. Findeis , Andrew Whiteley y John R. Krause. An unusual presentation of an uncommon lymphoma, hepatosplenic T-cell lymphoma. Proc (Bayl Univ Med Cent) . 2019 enero; 32 (1): 129–130.
Hematología y trasplante de médula ósea, Facultad de Medicina de la Universidad de Yale, New Haven, CT, EE. UU. Hepatosplenic T-Cell Lymphomas. Cancer Treat Res. 2019; 176:185-193.
|
Imágenes del caso
|
